FuNeL User Guide
Overview
FuNeL is a machine learning approach to generate functional networks using the co-prediction paradigm. Gene-gene functional interactions are defined by using a machine learning rule-based classification algorithm (BioHEL). This approach is based on the assumption that genes within the same classification rules have an increase likelihood to be functionally related. A general FuNeL protocol is shown below:
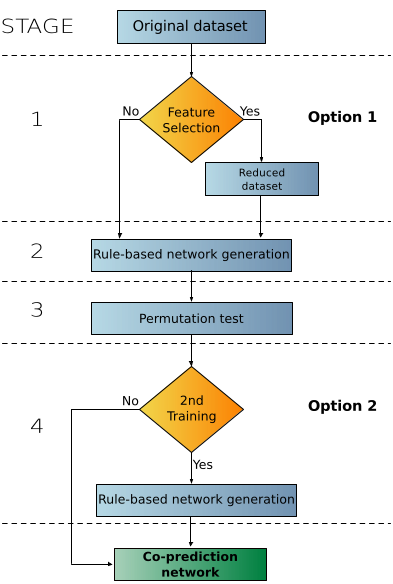
The rule-based machine learning build the model of the dataset that is later used to infer a network. It is statistically refined using permutation tests to filter out the non-significant nodes. The protocol has two optional settings: feature selection and 2nd stage of rule learning. It can be run with one of the following configurations:
- C1 (reduced dataset + 1 machine learning phase)
- C2 (original dataset + 1 machine learning phase)
- C3 (reduced dataset + 2 machine learning phases)
- C4 (original dataset + 2 machine learning phases)
Installation
System requirements
To run FuNeL a GNU/Linux or a Mac OS system is required with the following software installed:
- Weka (GNU General Public Licence)
- Python 2.7.x (Python Software Foundation Licence)
- NumPy (BSD Licence)
- GNU R (GNU General Public Licence)
Download
Before you start, you need to download FuNeL scripts.
Compilation
To run FuNeL, you need to compile BioHEL first. Its source code is distributed
along with FuNeL scripts. Use the Makefile provided in BioHEL folder:
tar axvf funel.tar.xz
cd funel/biohel
make
Configuration
FuNeL execution depends on three variables set in the coprediction.sh script.
- WEKA_PATH - path to the weka.jar file e.g.
weka-3.6.10/weka.jar
(FuNeL uses Weka's SVM-RFE implementation), - NUM_BIOHEL_RUNS - number of BioHEL runs, we suggest 10000 as default
- NUM_PERMUTATIONS - number of permuted datasets used for significance testing, we suggest 100 as default
Running FuNeL
To generate a co-prediction functional network run the following script from the main FuNeL directory:
./coprediction.sh <project_name> <dataset> <configuration> [<attributes>]
The script requires three parameters:
- project name - name of the results directory
- dataset - biological data in ARFF format
- configuration - protocol variant (number from 1 to 4), see the list in Overview
- attributes - number of attributes to retain from feature selection (only for configurations 1 and 3)
Example
The data directory contains a diffuse large B-cell lymphoma dataset in
ARFF format (from Shipp2002). To generate a co-prediction network
from this dataset with configuration C1 retaining 500 attributes, run:
./coprediction.sh lymphoma data/lymphoma_dataset.arff 1 500
The result file co-prediction_network.txt and other intermediate files will
be created in the results/lymphoma directory.
Post-filtering
If the dataset attributes are probes (instead of genes), you can still obtain
a gene functional network using the postfiltering.sh script.
It substitutes probe names with the correspondent gene names based on the
provided mapping file (see data/mapping.txt).
./postfiltering.sh <project_name> <mapping_file>
The mapping file is a two column tab separated file: probe_id gene_id. If two probes are mapped to the same gene the corresponding nodes are merged. If a probe name is not mapped to any gene name, the node is removed from the network.
Optimisation
The network generation process could be speeded up by parellisation of the
machine learning runs in coprediction.sh:
for run in `seq 1 $NUM_PERMUTATIONS`
do
scripts/rbng.sh $projectname 0 $NUM_BIOHEL_RUNS $run
done
The script rbng.sh creates the co-prediction network using a single permuted
data set. Due to datasets independence the machine learning phase could be
performed in parallel.
Contact
If you have any questions or comments about the FuNeL or this tutorial in particular, please contact us at jaume.bacardit.